 资源预览
资源预览




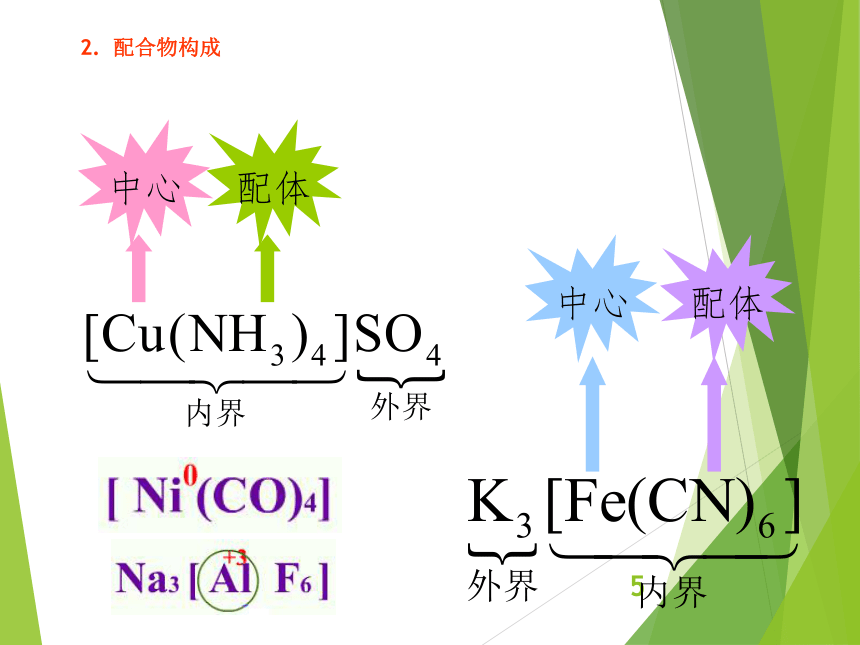
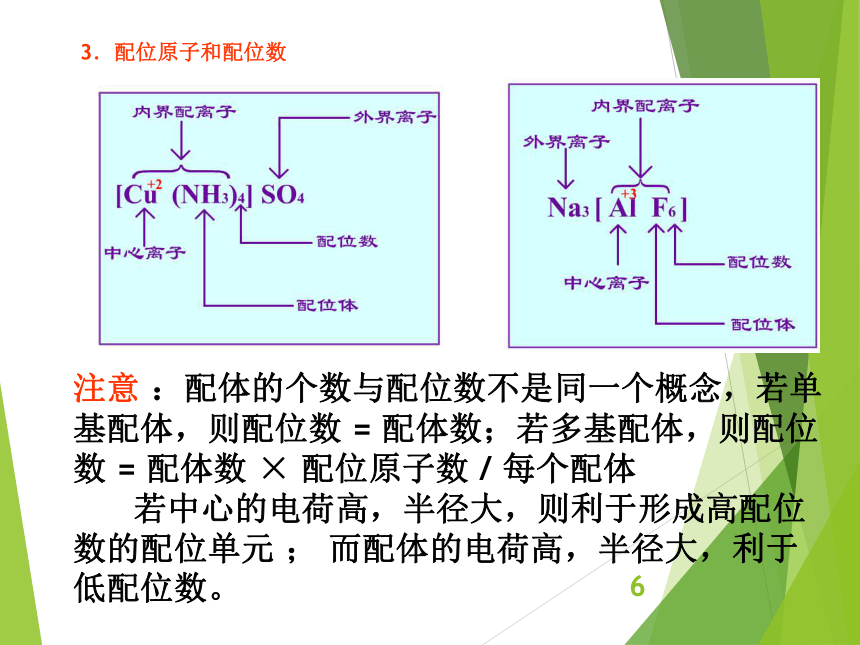
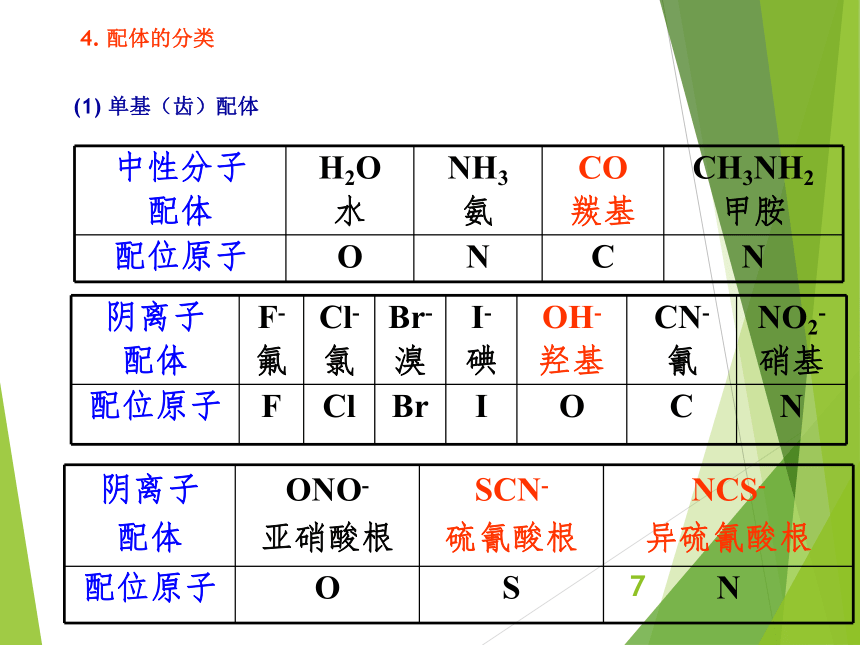
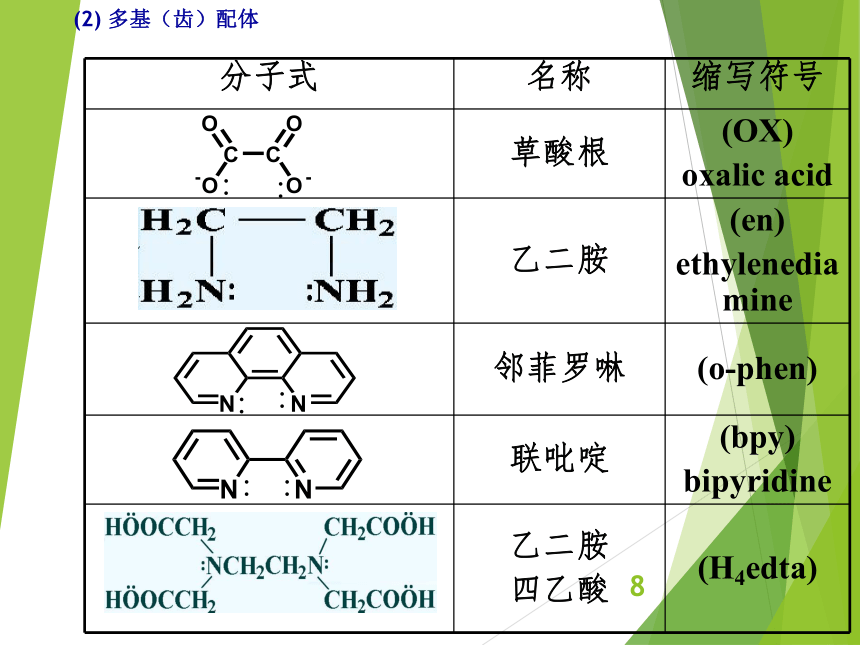
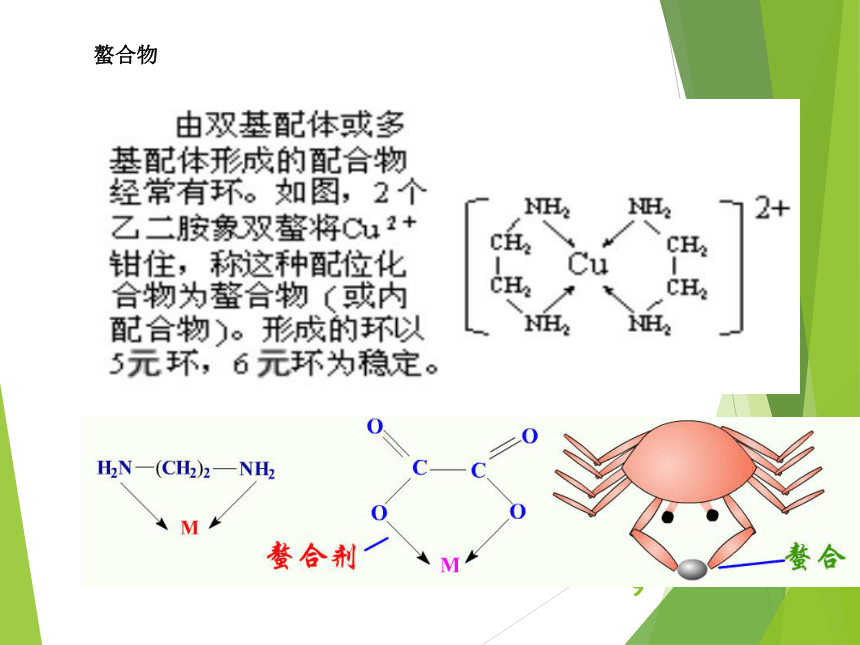
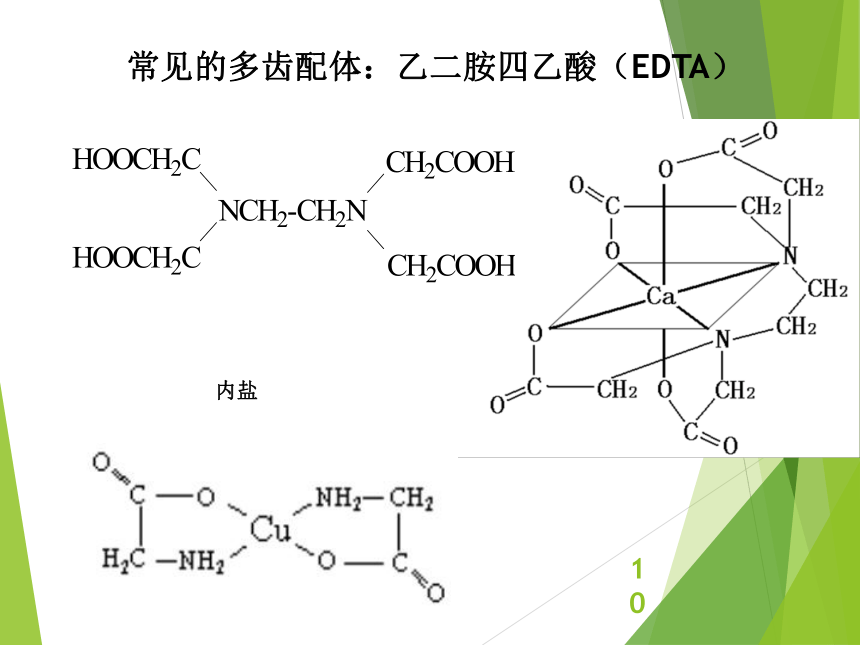
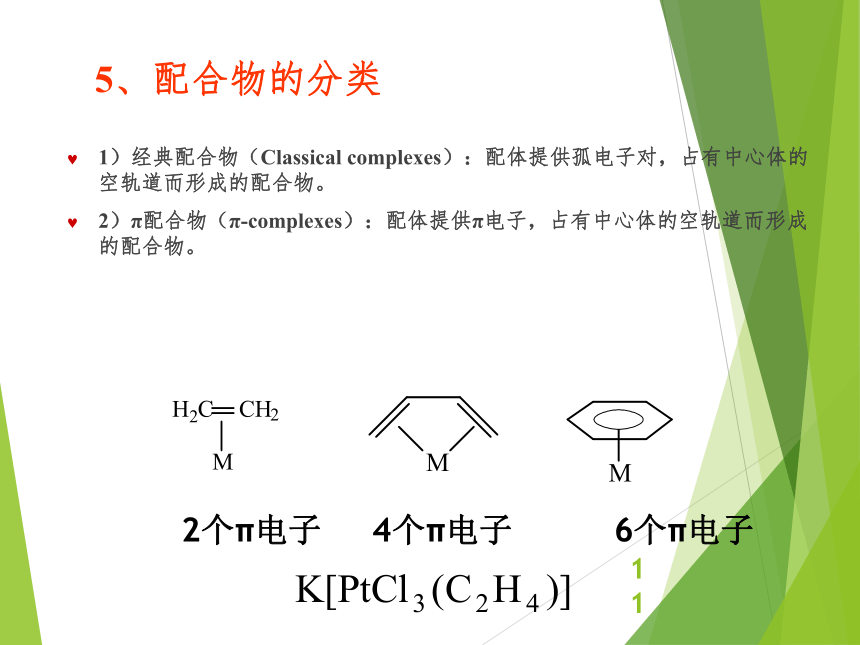

资源预览